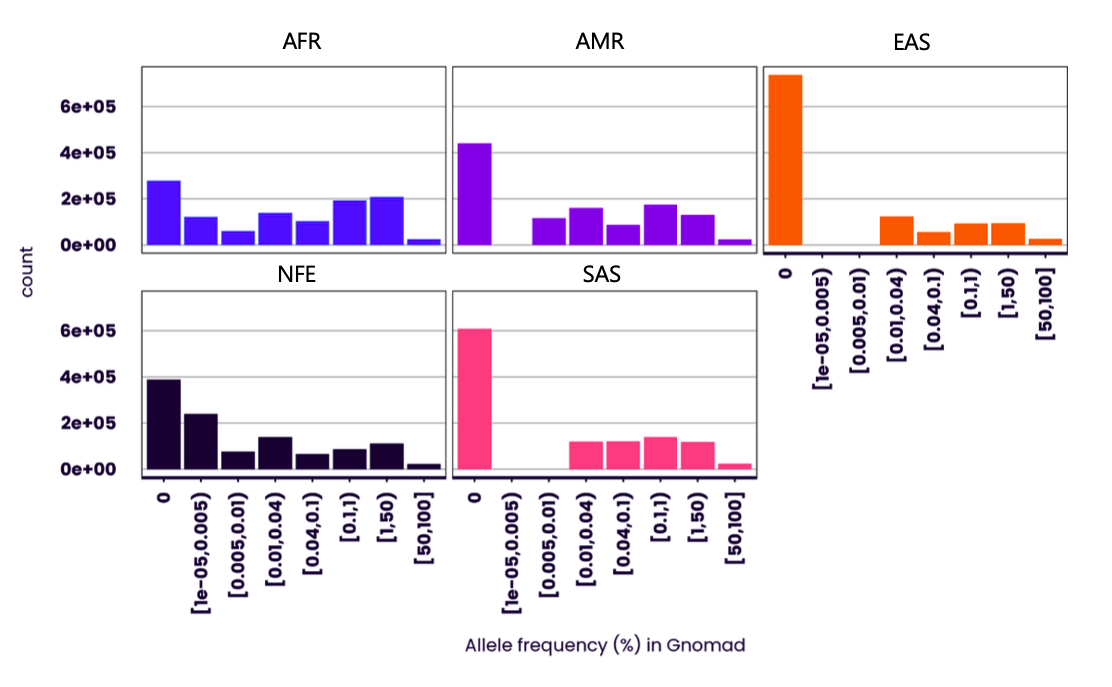
Figure 1 - Distribution of non-reference allele frequencies for chromosome 22 variants. Frequencies for chromosome 22 panel taken from Gnomad 4.0.0 database (1,873,149 variants). AFR: African/African American; AMR: Admixed American; EAS: East Asian; NFE: Non-Finnish European; SAS: South Asian.


